
在微生物表征的研究领域,开发用于生物体基因分型的分子生物学方法已成为一个核心方向。与依赖于生理或生长状态的传统方法不同,基因分型方法具有高度的特异性和灵敏度,并且基本不受微生物生理状态的影响。特别是聚合酶链式反应(PCR)及其衍生技术的发展,为在微生物无法培养的情况下进行检测和鉴定,提供了强有力的工具。
聚合酶链式反应(Polymerase Chain Reaction, PCR)是一种用于体外特异性扩增DNA片段的分子生物学技术。利用PCR及其相关技术,可以对实验室培养物以及环境样品中的微生物和高等生物进行快速检测与鉴定。
PCR扩增的DNA片段由引物的选择决定。引物是长度约20-30个核苷酸的人工合成的短DNA单链,其序列与待扩增DNA片段的起始和末端完全互补。这意味着,进行PCR的前提是必须预先知道目标DNA片段两端的序列。引物可以在实验室内合成,也可从商业供应商处购买。
PCR的基本流程包含三个核心步骤(见图1)。首先是变性(Denaturation),通过加热至90-96°C,使目标DNA双螺旋结构解开,分离成两条单链。第二步是杂交或退火(Annealing),在此阶段,引物会与其在DNA单链上的互补碱基结合。第三步是延伸(DNA Synthesis),DNA聚合酶以引物为起点,读取模板链并快速合成与之互补的新链。最终,一个DNA双螺旋分子变为两个,每个新分子都由一条原始链和一条新合成的互补链组成。通过周期性地重复变性、引物退火和DNA延伸这三个步骤,目标DNA片段得以指数级扩增。
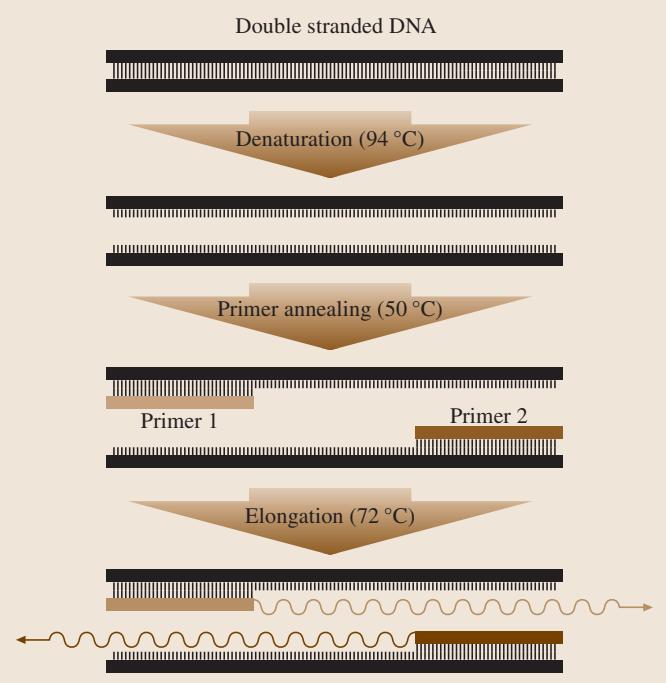
图1 PCR的三个基本步骤
随着针对特定菌群或物种的特异性引物的开发,PCR技术使得对培养物和环境样品中目标微生物的灵敏检测和快速鉴定成为可能。
扩增核糖体DNA限制性分析(Amplified Ribosomal DNA Restriction Analysis, ARDRA)技术,其核心操作是对PCR扩增的核糖体RNA(rRNA)基因进行限制性内切酶(能够识别并切割双链DNA上特定位点的酶)消化,然后通过电泳分离产生的片段。通过将得到的电泳图谱与数据库中的标准图谱进行比对,可以将分离株鉴定到物种水平,其分辨能力取决于所选用限制性内切酶的种类。
该方法能够快速筛选大量分离株,已在实验室培养物和自然基质中真菌的检测与鉴定方面获得了广泛应用。
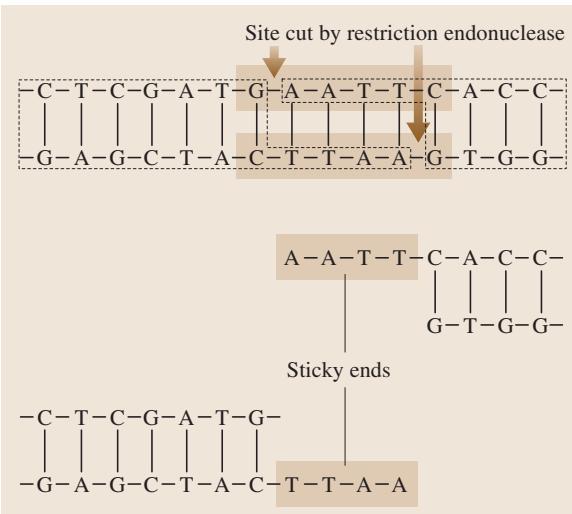
图2 真菌ARDRA技术流程示意图
核糖体DNA(rDNA)是该技术的研究靶点,因为它天然适合开发分类单元特异性引物。其结构中穿插着相对保守的区域(如18S rDNA、5.8S rDNA、28S rDNA基因)和非保守的区域(如ITS I、ITS II),并且在每个基因组中都有大量的拷贝数。实验中,研究人员使用通用引物(如ITS1和ITS4)或针对高等真菌(ITS1-F/ITS4-B)、担子菌(ITS1/ITS4-B)的特异性引物对来扩增rRNA基因及其插入的ITS序列。PCR产物经限制性内切酶消化后,根据酶切位点的不同,在凝胶电泳后会呈现出数量和大小各异的条带(见图2)。大多数酶产生的ARDRA图谱在物种水平上具有唯一性,其中,使用AluI、HpaII、HaeIII和TaqI等限制性内切酶进行消化,在区分腐朽菌方面被证明尤其有效。
随机扩增多态性DNA(Random Amplified Polymorphic DNA, RAPD),也称任意引物PCR(Arbitrarily Primed PCR, AP-PCR),是一种为那些序列信息不足、难以设计特异性引物的物种创建基因组DNA片段图谱(即DNA指纹)的方法。它主要用于鉴定(染色体)DNA上的菌株特异性变异(见图3)。
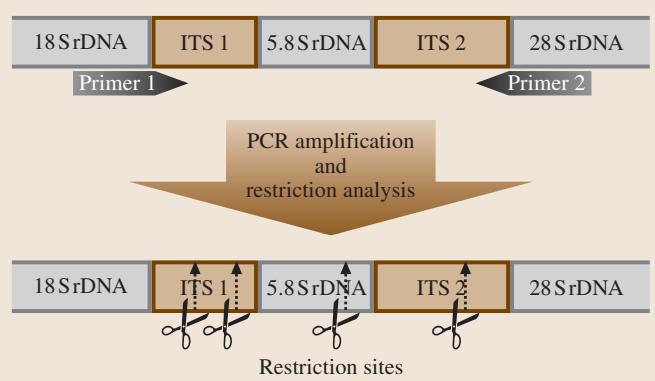
图3 使用相同随机引物对11种不同木材降解担子菌(Coniophora puteana)菌株进行RAPD分析得到的图谱。所有菌株最初被认为是相同的,并用于欧洲标准EN113。M:分子量标准品。
RAPD技术使用任意选择的短引物,这些引物会偶然地与基因组上某些位点完全或近似匹配,并以此为起点引发DNA合成,从而扩增出引物结合位点之间的DNA片段。通常,PCR在低严谨性条件下进行。不同菌株间引物结合位点的数量和位置各不相同,因此会产生菌株特异性的DNA指纹图谱。该技术的一个前提是,引物结合位点之间的距离必须在合理范围内,以确保DNA聚合酶能合成足够长的产物,使其包含另一个引物的退火位点。聚合酶从模板上“脱落”的速度,则取决于模板本身的纯度和组分(如GC含量)。如果您在实际工作中也面临类似的菌株鉴定与区分挑战,我们非常乐意与您一同探讨解决方案。
BRENDA方法主要用于表征原核生物,特别是细菌菌株。其流程是将染色体DNA用多种限制性内切酶消化,然后通过电泳分离DNA片段。由于不同菌株的基因组大小和酶切位点频率不同,会产生各异的片段图谱。但通常这些图谱非常复杂,难以分析。
而限制性片段长度多态性(Restriction Fragment Length Polymorphism, RFLP)方法则旨在减少可检测条带的数量。在该方法中,染色体DNA同样经过限制性酶切和凝胶电泳。随后,DNA片段被转移并固定在固相支持物上,如硝酸纤维素膜或尼龙膜。
通过杂交过程,可以检测到目标核苷酸序列。带有标记的DNA探针会与膜上互补的目标基因结合。由于待研究的两个菌株中限制性内切酶位点的存在与否不同,将导致含有目标基因的片段长度出现差异(见图4)。这种技术与BRENDA一样,能够有效区分不同菌株。
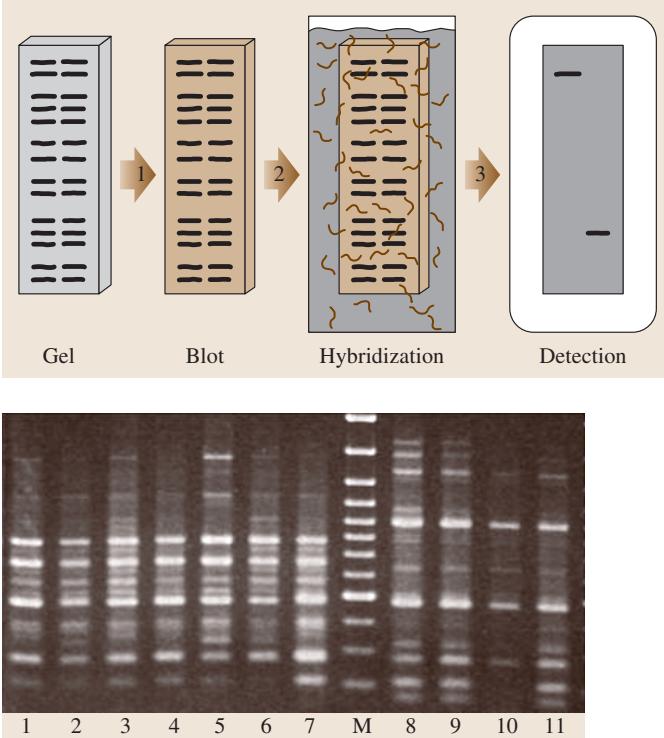
图4 RFLP方法示意图。(a) 两种待研究菌株的DNA经酶切后进行凝胶电泳分离,变性后转移(印迹)到尼龙膜或纤维素膜上。(b) 将印迹后的DNA与标记的DNA探针一同孵育,探针与膜上的互补目标基因结合。© 去除非特异性结合的探针后,可通过放射自显影等方式检测目标基因。
目前已有多种核酸探针的标记和检测系统。放射性标记的探针通过放射自显影进行可视化;非放射性系统则基于酶促、光化学或化学方法将报告基团(如荧光染料、与化学发光检测或银增强耦合的标记酶)引入探针,可通过光学、发光、荧光或金属沉淀检测系统进行高灵敏度检测。
由于rRNA基因在不同物种间具有一些高度保守的区域,这使得来自一种生物(如大肠杆菌)的rRNA可以用作通用探针,因此这些基因被常规用作靶基因。这种被称为“核糖体分型(Ribotyping)”的技术,可视为一种特殊的RFLP,用于检测不同菌株间rRNA基因限制性图谱的差异。通常,其图谱比靶向其他基因的RFLP更复杂,因为rRNA基因在基因组中以多拷贝形式存在。核糖体分型的分辨能力取决于所研究的物种和选择的限制性内切酶。
商业化的全自动系统,如Ribo-Printer微生物鉴定系统,极大地推动了核糖体分型的应用。这种分子工作站能够自动完成染色体DNA的限制性酶切(使用EcoRI或其他酶)、凝胶电泳分离限制性片段,并同时将DNA片段印迹到膜上。DNA片段与基于核糖体rRNA操纵子基因保守区域的细菌探针进行杂交。每个指纹图谱都存储在数据库中,以便未来进行比较和鉴定。Ribo-Printer系统在质量控制和菌种鉴定领域应用频繁。要获得稳定可靠的图谱,对实验操作的每一步都有极高的要求,这正是专业检测实验室的核心价值所在。
精工博研测试技术(河南)有限公司(原郑州三磨所国家磨料磨具质量检验检测中心),央企,国字头检测机构,专业的权威第三方检测机构,专业检测微生物基因分型与鉴定,可靠准确。欢迎沟通交流,电话19939716636
此外,针对特定生理活动(如硝化作用、固氮作用、毒力相关基因等)的基因探针,在表征特定功能的微生物亚群或微生物群落时非常有用,尤其是在群落的分类学或物种组成并非主要关注点的情况下。
荧光原位杂交(Fluorescence in situ Hybridization, FISH)主要用于原核生物群落的研究。它允许在微生物的自然微生境中对其进行直接鉴定和定量,无论是针对特定的分类群还是广谱的微生物群体。由于是对完整细胞进行杂交,该方法避免了因DNA提取、PCR扩增和克隆等步骤可能引入的偏差。FISH不仅是研究种群中个体的强大工具,也具备研究种群动态和追踪释放到环境中的微生物的潜力。
复杂的微生物群落组成通常通过靶向rRNA的核酸探针进行分析:将完整细胞固定,其16S或23S rRNA在严谨条件下与荧光标记的分类单元特异性寡核苷酸探针进行杂交。标记后的细胞通过荧光显微镜进行观察。在灵敏度方面,扫描共聚焦激光显微镜(SCLM)优于落射荧光显微镜,并能同时评估多个分类群的分布情况。
大多数细胞中rRNA含量丰富,以及用于比较序列分析的庞大rRNA数据库的可用性,是靶向rRNA的核酸探针的主要优势。借助ARB等软件包,可以轻松设计rRNA寡核苷酸探针。用户可以选择特定的生物或类群,定义探针长度、G+C含量和靶区域等参数,ARB的探针设计工具便会在整个序列数据集中搜索潜在的靶位点。由于ARB数据库会频繁更新,使用旧探针前应重新核对数据库。
对于没有共同诊断靶位点的微生物类群,应使用多个探针进行检测。为了提高灵敏度,例如在检测和追踪功能基因时,建议使用辣根过氧化物酶标记的寡核苷酸探针。市面上也可以购买到用多种荧光染料标记的寡核苷酸探针。
变性梯度凝胶电泳(Denaturing Gradient Gel Electrophoresis, DGGE)近年来被广泛用于分析微生物群落的构成。当需要分析种群结构的时空动态时,该方法尤其有用。它能够分离大小相同但核苷酸序列不同的DNA片段,其分离依据是片段的变性特性。在微生物群落表征中,通常直接从总DNA样品中通过PCR获得核糖体RNA基因,然后进行DGGE分析。最终得到的条带图谱可作为该微生物群落的指纹。
在含有递增浓度变性剂(如尿素)的凝胶中,或在递增的温度梯度中(即温度梯度凝胶电泳,TGGE)进行电泳时,DNA分子在到达其熔解点(即特定变性剂浓度或温度)之前保持双链结构。一旦熔解,DNA分子会分支,从而在凝胶中的迁移率降低。由于熔解行为主要由核苷酸序列决定,理论上,混合模板DNA中存在的任何rDNA基因都可以被特异性扩增并在DGGE凝胶上得到分离。DGGE电泳后,rDNA片段可以被切胶回收、测序,并与公共数据库中的已知序列进行相似性分析。需要注意的是,凝胶上的一个条带往往不只代表一个物种,因此该方法更适用于复杂度较低的群落。
该技术也可能受到DNA提取效率的限制,不同微生物和不同环境类型(土壤、淤泥、水体)的提取效率存在差异。此外,研究表明,当模板丰度差异较大时,会发生扩增偏好,丰度较高的序列会被优先扩增。












 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
