
电子探针X射线微分析 (EPMA)大多数固体物质在显微尺度上由化学差异化的微观结构特征,其特征尺寸在微米到纳···
大多数固体物质在显微尺度上由化学差异化的微观结构特征,其特征尺寸在微米到纳米范围内。许多物理、生物和技术过程在宏观尺度上受显微尺度上的化学过程控制。电子探针X射线微分析仪 (EPMA) 是一种基于扫描电子显微镜 (SEM) 的分析工具,使用精细聚焦的电子束激发样品发射特征性X射线。
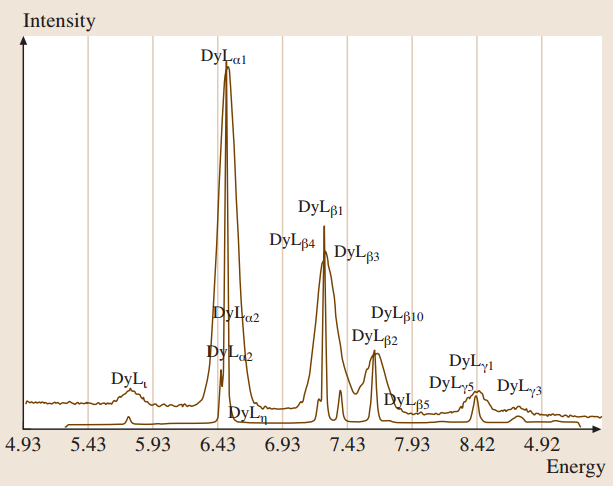
EPMA/SEM 能够定量分析主要、次要和微量元素组成,除了氢、氦和锂外,浓度低至大约10-5 的质量分数。该技术一般被认为是非破坏性的,通常应用于平坦、金相抛光的样品。SEM 允许将该技术应用于特殊情况,例如粗糙表面、颗粒、基板上的薄层和无支撑的薄层。此外,SEM 提供全范围的形态成像和结构晶体学能力,使其能够在微米到纳米尺度上表征形貌、表面层、横向成分变化、晶体取向以及磁场和电场。两种不同类型的X射线光谱仪广泛使用,能量色散光谱仪 (EDS) 和波长色散(或晶体衍射)光谱仪 (WDS)。这些光谱仪的特性是高度互补的:一个的弱点通常被另一个的优势所弥补。因此,它们通常在同一个电子束仪器上一起使用。最近,硅漂移探测器 (SDD) 的出现将EDS输出计数率扩展到100 kHz到500 kHz范围内。下图展示了Dy L系列一部分的EDS和WDS光谱的比较,显而易见,WDS的光谱分辨率显著优于EDS。
20 keV束流激发镝l族X射线的EDS和WDS比较
EDS和WDS X射线光谱仪特性比较
特性 EDS(半导体) WDS 能量范围 0.1–25 keV (Si)
0.1–100 keV (Ge)0.1–12 keV (4晶体) 在MnKα处的分辨率 130 eV (Si); 125 eV (Ge) 2–20 eV (E, 晶体) 瞬时能量覆盖范围 全范围 分辨率,2–20 eV 死时间 50 微秒 1 微秒 固体角(立体弧度) 0.05 – 0.2 0.01 量子效率 ≈ 100%,3 keV到15 keV (Si) < 30%,可变 最大计数率,EDS ≈ 3 kHz(最佳分辨率) 100 kHz(单光子能量) 最大计数率,SDD ≈ 30 kHz(映射)
≈ 15 kHz(最佳分辨率)
≈ 400 kHz(映射)全光谱采集时间 10秒到200秒 600秒到1800秒 特别优势 在所有位置进行定性分析时查看完整光谱 解决峰值干扰;快速脉冲
定性分析,即识别谱中特征峰对应的元素,对于主要成分(例如浓度大于0.1%质量分数的元素)通常是直接的,但对于次要(0.01-0.1%质量分数)和痕量(<0.01%质量分数)成分则可能具有挑战性。这对于EDS光谱尤其如此,当次要和痕量成分峰位于主要成分峰的附近(<100 eV 之内)时,这些干扰需要进行峰值解卷积,尤其是在次要或痕量元素是轻元素 (Z < 18) 时,对于EDS而言可能只有一个峰可解析。在这种情况下,自动化的计算机辅助EDS定性分析必须始终进行手动检查以确保准确性。WDS 的优越光谱分辨率通常能够在这种情况下分离主要/次要或主要/痕量峰,并且不会受到EDS的光谱伪影(如叠加峰和逃逸峰)的影响。然而,使用WDS时必须格外小心,以避免错误地将布拉格衍射方程中的高阶反射 (n = 2, 3, 4, ...) 误解为其他元素的峰。
定量分析分三个阶段进行:(1)提取峰强度;(2)标准化;(3)计算基体效应。对于EDS光谱,首先通过应用背景模型或数学滤波去除背景。然后通过多线性最小二乘 (MLLSQ) 方法进行峰值解卷积。MLLSQ 需要在用户仪器上针对每个元素的峰形模型,这些模型不受其他成分干扰。从包含两个或更多成分贡献的未知样品的峰区域通过构建所有成分的参考峰形的线性组合进行解卷积。合成峰与测量光谱进行比较,直到根据统计标准(例如逐通道最小化卡方)获得最佳匹配。对于WDS光谱,分辨率通常足以分离峰干扰,因此唯一的问题是去除背景。由于背景在WDS峰的狭窄能量窗口内线性变化,因此可以通过在峰的两侧进行背景测量进行准确的背景校正。
准确定量电子探针X射线微分析的基础是测量未知样品中X射线峰的强度与标准样品中相同峰的强度之比,所有测量均在相同的束能、已知电子剂量(束电流 × 时间)和光谱仪效率下进行。这个比值称为k值,与样品和标准中元素的质量浓度比值成正比:
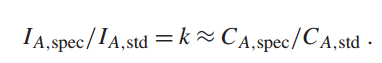
这个标准化步骤定量消除了探测器效率的影响,并减少了基体校正所需的许多物理参数的影响。EPMA的一个很大优势是所需标准套件的简单性。纯元素和简单的化学计量化合物(对于那些在真空中不稳定的元素,如硫的硫铁矿,FeS2)已经足够了。这是一个巨大的优势,因为在微米尺度上制备均匀的多元素混合物通常由于相分离而非常困难。
k值与(样品中A元素的浓度/标准中A元素的浓度)之间不是相等的,因为基体或元素间效应的作用。也就是说,元素B的存在在生成、传播和检测时会改变元素A的强度。幸运的是,这些基体效应的物理起源是众所周知的,通过基础物理学以及经验测量,已经开发出了原子序数效应 (Z)、吸收效应 (A) 和荧光效应 (F) 的乘法校正因子:
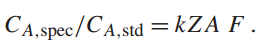
显然,所有这三个基体效应 - Z、A 和 F - 都强烈依赖于所测样品的成分,这就是我们要解决的未知数。基体效应的计算必须从样品浓度的初步估计开始,迭代进行,直到得到最终的计算值。测得的k值用于提供样品成分的初步估计,方法是将浓度设为归一化的k值:
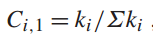
然后使用初始浓度值计算一组初始基体校正值,再用这些基体校正值计算预测的k值。将预测的k值与实验值进行比较,如果值在定义的误差范围内一致,则计算终止。否则,循环重复。通常在三次迭代内收敛。
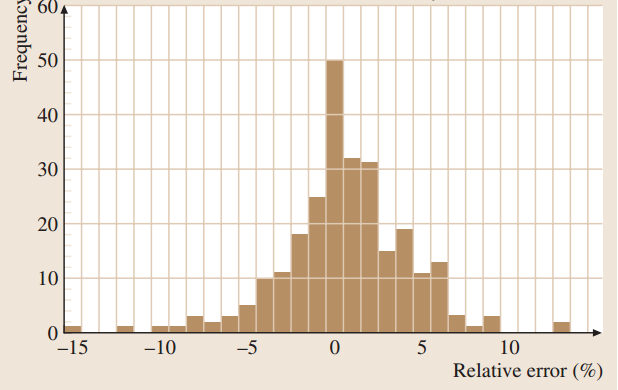
这种基体校正程序在过去25年中通过使用各种已知成分的微均匀材料作为测试样品,包括合金、矿物、化学计量的二元化合物等反复测试。下图显示了二元合金相对于纯元素标准的相对误差(定义为[测量值-真实值]/真实值×100%)的典型分布。
呈现X射线微分析信息的一种强大方法是以成分图或图像的形式,描绘元素成分的区域分布。这些图可以与提供形态信息的SEM图像同时记录。WDS、EDS或SDD在定义的X射线光子能量范围内的数字输出记录在每个扫描图像的像素上。成分映射的最高级别涉及为扫描图像的每个像素收集一个光谱,或至少一些光谱强度窗口。这些光谱数据随后经过背景校正、峰值解卷积、标准化和基体校正,以实现定量分析。生成的图实际上是局部浓度的记录,当显示时,灰度或颜色比例实际上与浓度相关。
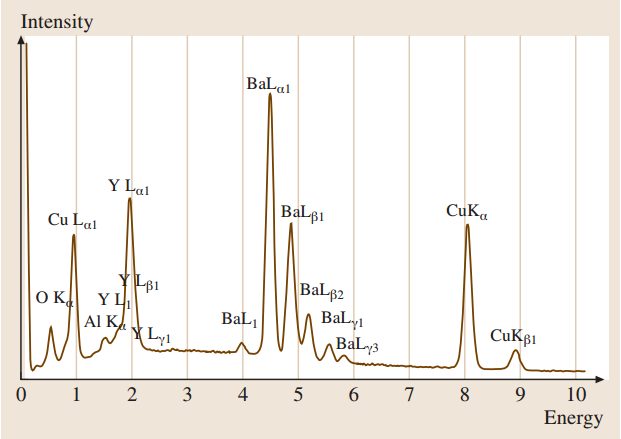
多组分样品YBa2Cu3O7的典型EDS光谱
下图展示了铝-镍合金的成分图例子。

铝镍(Ni - Al)合金的成分图(Ni, Al和Fe)和SEM图像(背散射电子,BSE)显示出一个复杂的微观结构,在不连续相中分离出少量的铁成分
请联系我们,为您提供快速、优质的服务!
可以根据您的需求,定制检测。
如果您不满意检测结果,我们可以采用多种方式,直到您满意为止。
联系我们的工程师,为您做专业解答。
一对一为您答疑解惑













 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
